手性叔丁基亚磺酰胺的发现历程与合成方法。这种试剂最早由美国加州大学伯克利分校(University of California, Berkeley)的Jonathan A. Ellman教授团队设计,作为一种手性辅基,可用于高效构建手性胺结构。接下来我们就从制备手性亚胺说起,具体聊聊其在有机合成反应中的应用。
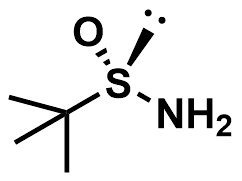
(S)-(-)-叔丁基亚磺酰胺的分子结构
此前我们也提到,构建手性胺结构可以从手性的亚胺出发,使用亲核试剂对其非对映选择性加成来实现。理论上讲,手性叔丁基亚磺酰胺与醛、酮等羰基化合物脱水缩合便可制得相应的手性亚胺,但在这种试剂问世以前,人们只能利用其他手段达到这一目的。1995年,西班牙马德里自治大学(Universidad Autόnoma de Madrid)的García Ruano教授团队设想以亚磺酰亚胺作为原料,借助氮原子修饰的辅基进行手性诱导,实现其不对称亚甲基化,最终得到光学纯的吖丙啶。他们从手性的对甲苯亚磺酰亚胺出发,以锍叶立德作为亚甲基化试剂,[1+2]加成后消除手性辅基便可得到目标产物。但这种方法得到的[1+2]加成产物非对映选择性不够理想,好在可以通过柱层析分离两种异构体,随后再消除保护基分别得到两种构型的吖丙啶。
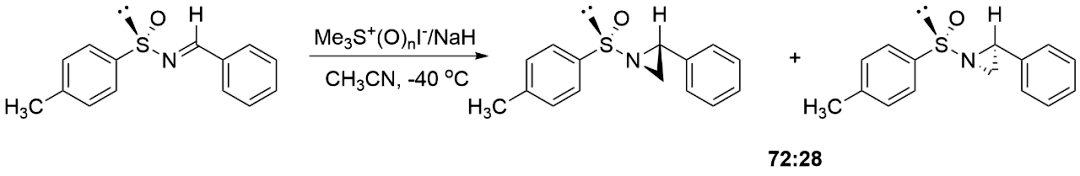
手性对甲苯亚磺酰亚胺的不对称亚甲基化(图片来源:参考资料[1])
第二年,该团队发现将亚胺修饰的手性对甲苯亚磺酰基换作叔丁基亚磺酰基时,反应的非对映选择性得到了明显的改善。相应的亚胺可使用光学纯的双丙酮-D-葡萄糖(DAG)作为手性诱导试剂,在碱的作用下与叔丁基亚磺酰氯缩合得到非对映异构体纯的亚磺酸酯,柱层析分离提纯后参考Franklin A. Davis教授发展的合成对甲苯亚磺酰亚胺的方法,以LiHMDS作为氮亲核试剂对其亲核取代,原位形成手性的N,N-双(三甲基硅基)叔丁基亚磺酰胺,再进一步与醛脱水缩合来合成。改变第一步反应的碱和溶剂可以分别获得两种差向异构体。
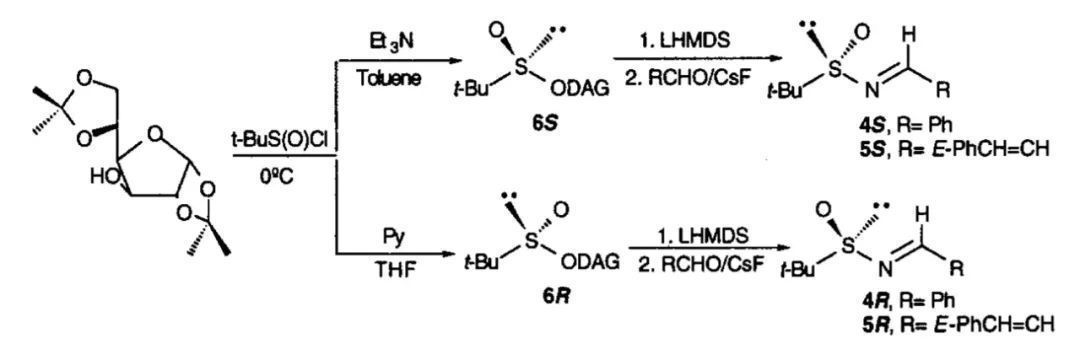
早年手性叔丁基亚磺酰亚胺的制备方法(图片来源:参考资料[2])
Jonathan A. Ellman教授成功合成并分离出手性叔丁基亚磺酰胺则为这一领域的研究带来了重要的改变,其实际意义不只是将两步反应缩短为一步,同样标志着手性亚磺酰亚胺合成策略的突破。除此之外,手性叔丁基亚磺酰胺已实现商品化,人们可利用这种试剂轻松获取目标亚胺产物,合成效率大大提高。
当然,他们还进一步对合成手性亚磺酰亚胺的反应条件进行了全面的考察。对于不同类型的羰基化合物,反应需借助不同的Lewis酸促进叔丁基亚磺酰胺与底物脱水缩合。制备醛衍生的亚胺可以使用CuSO4作为Lewis酸及脱水剂,以叔丁基亚磺酰胺作为限制试剂,此时醛的使用量最少(1.1当量),而当醛的反应活性较低或制备酮衍生的亚胺时,Ti(OEt)4参与反应的效果更好。缩合后硫原子中心的手性构型不会发生改变,因而可以得到光学纯的亚胺产物。叔丁基亚磺酰亚胺化学性质较为稳定,对水汽及空气不敏感,无需在无水无氧的氛围下操作。不过,这类化合物长时间处于室温下或光照会发生少量分解,作者建议将其置于-5 ℃下密闭储存。
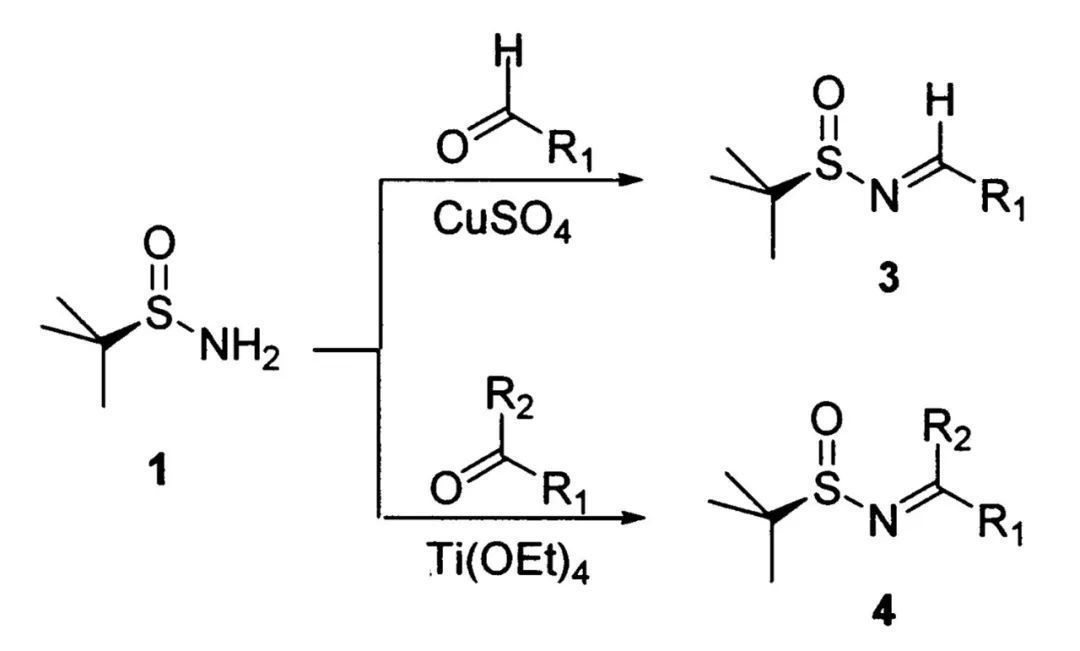
手性叔丁基亚磺酰胺与醛、酮缩合形成相应的亚胺(图片来源:参考资料[3])
得到手性的叔丁基亚磺酰亚胺,人们便可以利用不同的亲核试剂对其亲核加成得到各种结构的胺,如格氏试剂、有机锂试剂对醛衍生的亚胺加成,随后加入HCl的MeOH溶液消除叔丁基亚磺酰基保护基,能以优异的选择性得到相应的手性α-支链伯胺盐酸盐。该方法不仅适用于芳香亚胺,脂肪族亚胺也能顺利参与反应,即便其α-H酸性较强也不会受到明显影响。而使用NaBH4作为还原剂,酮衍生的亚胺也可非对映选择性还原为相应的手性α-支链胺,殊途同归。
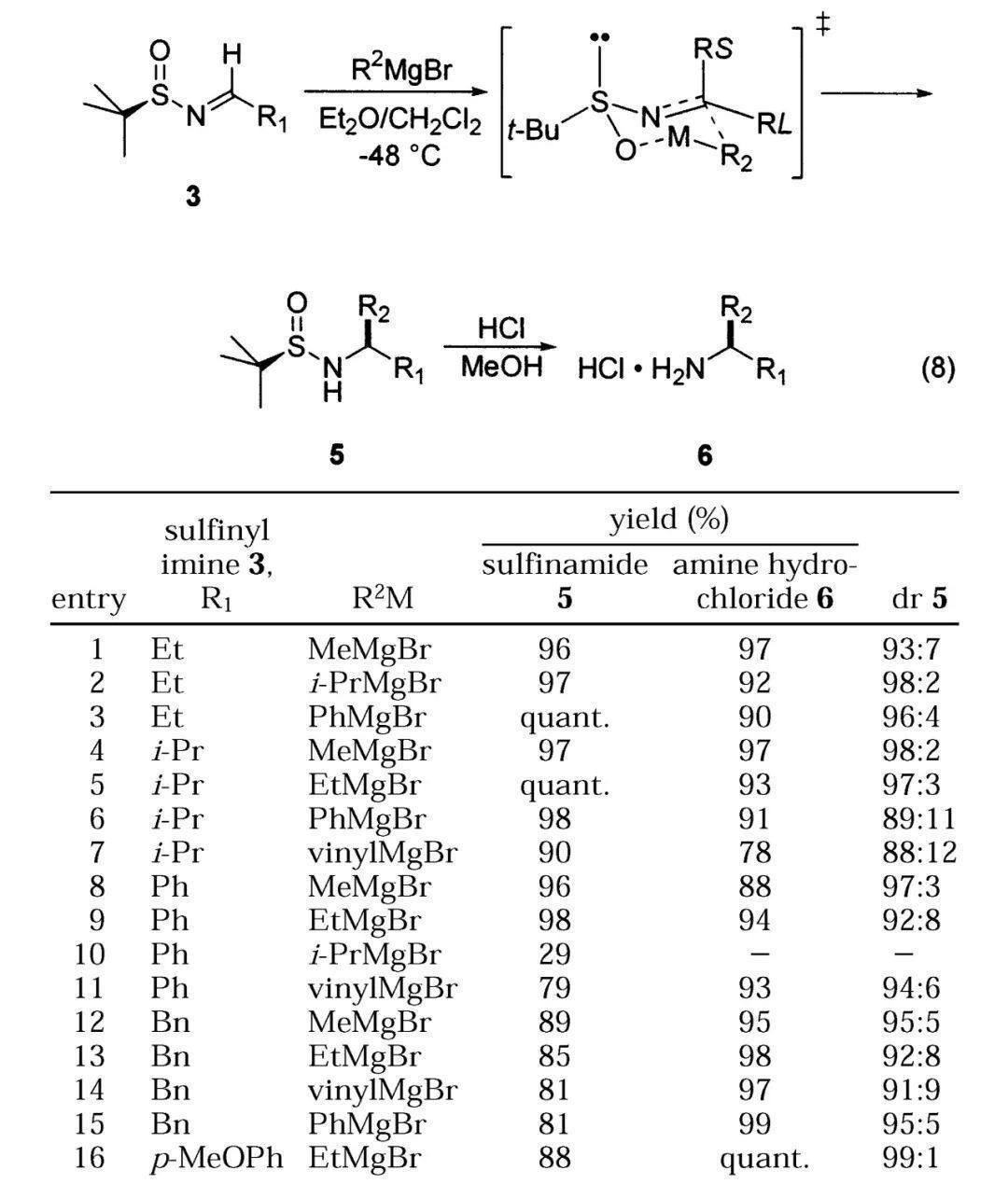
格氏试剂对醛衍生的手性叔丁基亚磺酰亚胺亲核加成制备手性的α-支链伯胺(图片来源:参考资料[3])

酮衍生的手性叔丁基亚磺酰亚胺还原制备手性的α-支链胺(图片来源:参考资料[3])
此外,使用Me3Al作为活化剂,酮衍生的亚胺还可与有机锂试剂反应,得到手性的三级甲胺。这种结构在复杂的天然产物及药物分子中较为常见,但一直苦于缺少通用的策略来实现,手性叔丁基亚磺酰胺的出现可谓帮了大忙。

以酮衍生的手性叔丁基亚磺酰亚胺作为原料合成手性的三级甲胺(图片来源:参考资料[3])
这种试剂不仅可以用于合成手性胺,还可实现手性氨基酸、氨基醇等其他包含氨基结构的构建。Ellman教授以甲酸酯作为原料,将其α位攫氢得到相应的烯醇化锂,后者与ClTi(OiPr)3转金属化制得烯醇化钛中间体,与叔丁基亚磺酰亚胺混合能以优异的立体选择性得到β-氨基酸。早年人们也发展了其他的方法来合成β-氨基酸,如Arndt-Eistert同系化、胺作为亲核试剂对丙烯酸酯Michael加成,但随着取代基增多、结构更加复杂,以上方法则不再适用。Jonathan A. Ellman教授发展的方法突破了这一局限,甚至能用于合成空间位阻较大的α,α,β,β-四取代β-氨基酸。
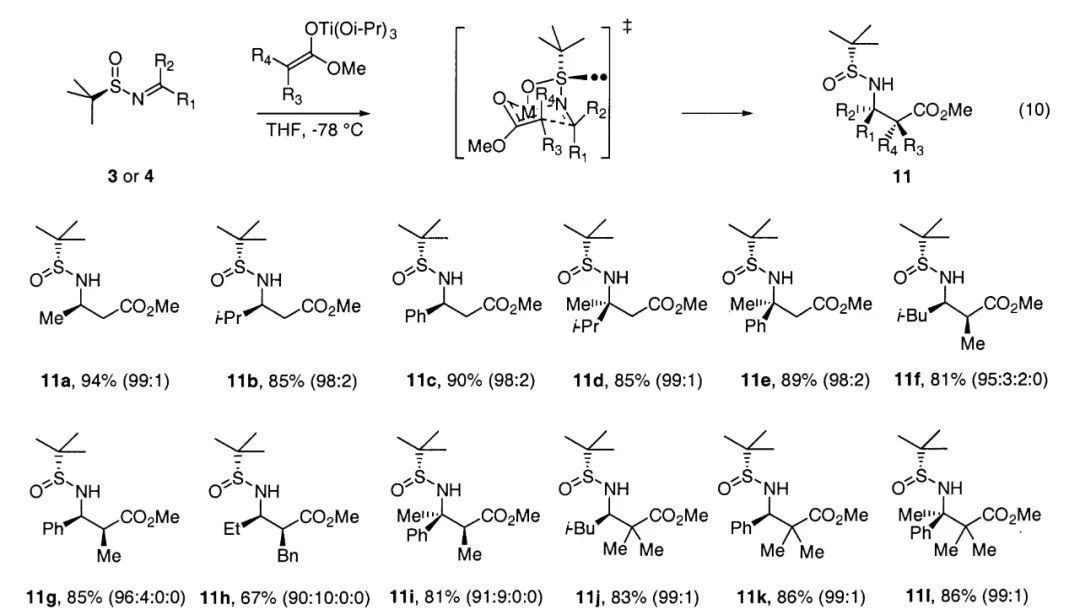
从手性叔丁基亚磺酰亚胺出发合成β-氨基酸(图片来源:参考资料[3])
美国天普大学(Franklin A. Davis)的Franklin A. Davis教授还从乙醛酸酯出发,手性叔丁基亚磺酰胺与醛羰基缩合后,再以格氏试剂或有机锌试剂作为亲核试剂与之反应,可以得到一系列的手性α-氨基酸。体系中需加入BF3·OEt2作为Lewis酸活化亚胺,促进亲核加成过程。
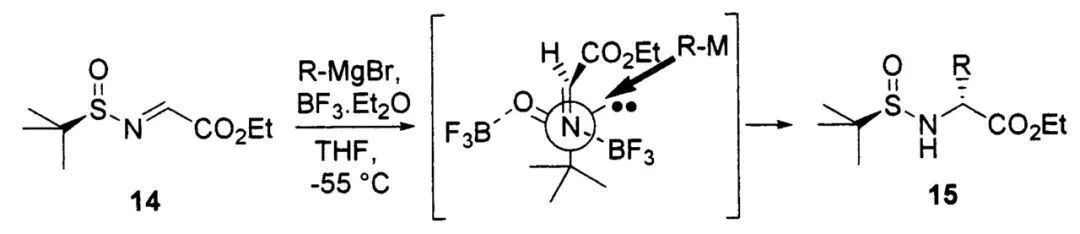
从手性叔丁基亚磺酰亚胺出发合成α-氨基酸(图片来源:参考资料[3])
讲到这里,想必大家已经猜到了手性氨基醇的合成方法。人们只需预先制备相应醛衍生的α-氧代叔丁基亚磺酰亚胺,再使用格氏试剂对其亲核加成,随后消除氮、氧原子的保护基便能得到手性1,2-氨基醇。
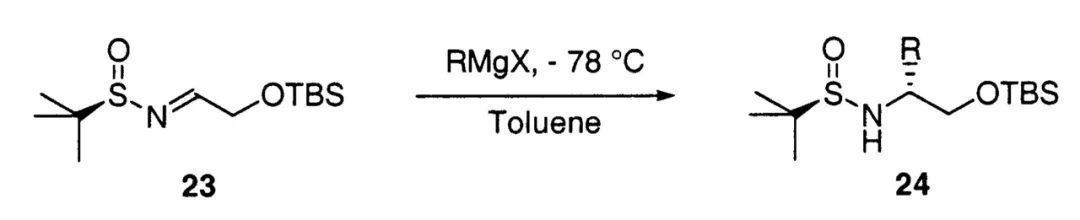
以手性叔丁基亚磺酰亚胺作为原料合成手性1,2-氨基醇(图片来源:参考资料[3])
1,3-氨基醇的合成思路与此类似,Ellman教授设计了一种简单的方法构建β-羟基叔丁基亚磺酰亚胺,芳香乙酮衍生的亚胺在LDA的作用下α位去质子化形成烯胺中间体,随后对醛亲核加成即可。
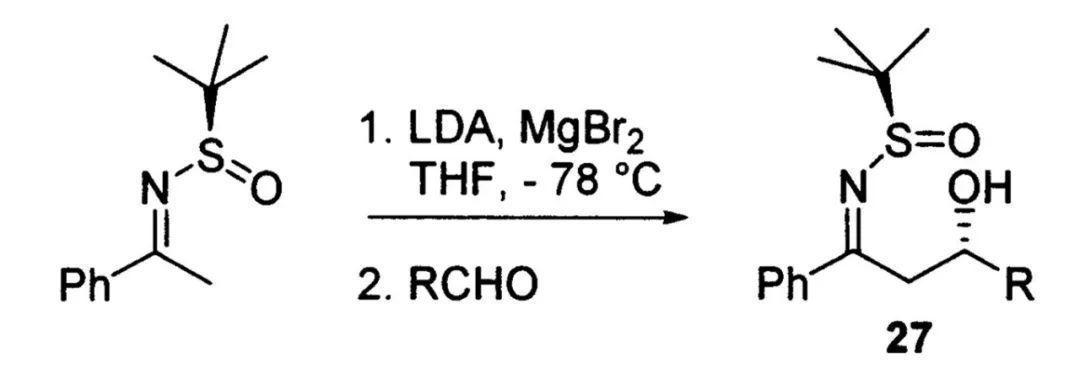
制备β-羟基叔丁基亚磺酰亚胺(图片来源:参考资料[3])
除了用作手性辅基,手性叔丁基亚磺酰胺还可用于合成手性配体,与Lewis酸催化剂结合,在不对称催化反应中发挥作用。例如,Ellman教授便设计了如下双亚磺酰亚氨基胺配体,结合Cu(SbF6)2可以完成对映选择性的Diels-Alder反应。
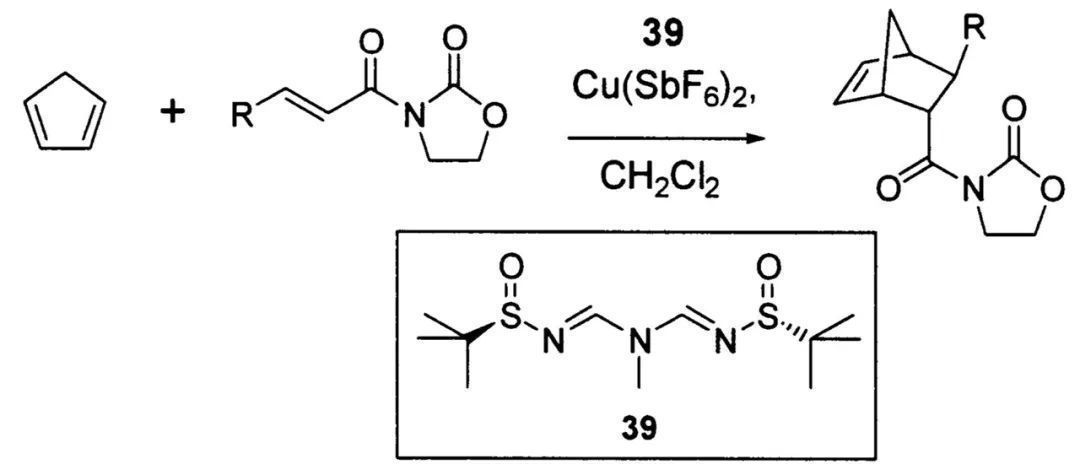
手性叔丁基亚磺酰胺用于合成手性配体(图片来源:参考资料[3])
随着金属有机化学的发展,手性叔丁基亚磺酰亚胺参与的过渡金属催化反应自是不会缺席,受篇幅限制,在此不作赘述。不过,看罢以上内容,且不要以为合成这种化合物十分简单,脱水缩合的效果会明显受到底物结构、亲核试剂及溶剂种类等诸多因素的影响,其研究也经历了曲折的发展过程,欲知详情,下回分解。
参考资料
[1] JoséL García Ruano et al., On the reaction of chiral sulfinimines with sulfur ylides: a novel route to the asymmetric aziridination. Tetrahedron Lett. 1995, 36, 295.[2] JoséL García Ruano et al., Asymmetric aziridination by reaction of chiral N-sulfinylimines with sulfur ylides: Stereoselectivity improvement by use of tert-butylsulfinyl group as chiral auxiliary. Tetrahedron: Asymmetry 1996, 7, 3407.[3] Jonathan A. Ellman et al., N-tert-Butanesulfinyl Imines: Versatile Intermediates for the Asymmetric Synthesis of Amines. Acc. Chem. Res. 2002, 35, 984.[4] MaryAnn T. Robak et al., Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600.[5] Tony P. Tang et al., The tert-Butanesulfinyl Group: An Ideal Chiral Directing Group and Boc-Surrogate for the Asymmetric Synthesis and Applications of β-Amino Acids. J. Org. Chem. 1999, 64, 12.[6] Takuya Kochi et al., Asymmetric Synthesis of syn- and anti-1,3-Amino Alcohols. J. Am. Chem. Soc. 2002, 124, 6518.


目前评论: