- A+
聚烯烃弹性体催化剂研究进展
王 静1* ,史永森2 ,李亚玲1 ,梁 斌1 ,许 胜2*
1. 兰州石化职业技术学院,甘肃 兰州 730060
2. 华东理工大学 化学与分子工程学院,上海 200237
摘要: 聚烯烃弹性体( POE) 是一类高性能、高附加值的聚烯烃材料。随着其应用领域的不断拓展,市场份额迅速扩大,国内各大相关企业纷纷投入巨资进行研究开发。POE 催化剂作为核心技术,目前主要控制在西方公司的手中,因此开发具有自主知识产权的催化剂,是当前 POE 领域研究的热点。
为了给国内同行提供参考,课题组结合国内外的研究工作,对目前主流的 POE 催化剂—限定几何构型茂金属催化剂( CGC) 、桥连双配体结构茂金属催 化剂和苯氧基亚胺催化剂( FI) 的发展进行了评述,对近 5 年这 3 类催化剂的微观结构、催化活性以及结构与催化性能的关系,特别是对催化乙烯/1-辛烯共聚反应中的催化剂活性以及 1-辛烯插入率进行了详细地比较,考察了不同的有机配体对催化剂催化行为、聚合物组成的影响,揭示了均相催化条件下通过改变配体结构以提高聚合反应温度和 1-辛烯插入率的技术。
我们摘录了 67 篇文献,涉及 109 种催化剂,相信我们的工作对于国内同行设计、合成新型 POE 催化剂具有重要的参考作用。
关键词: 聚烯烃弹性体; 茂金属催化剂; 苯氧基亚胺催化剂; 进展
聚烯烃材料是目前用途最广泛、产量最大的一类高分子材料,通常是指由乙烯与丙烯和 α-烯烃 ( 通常指 1-丁烯、1-戊烯、1-己烯、1-辛烯等) 及某些环烯烃均聚或共聚而得到的一类树脂材料。在乙烯的聚合反应中引入 α-烯烃不仅降低聚合物的密度和结晶度,而且可调节聚合物的熔融指数,改善聚合物的加工性能。
随着共聚物中 α-烯烃含量的提高,材料的性能从聚烯烃塑料向聚烯烃弹性体 ( Polyolefin elastomer,简称 POE) 转变,POE 通常由茂金属、镍系、钒系等催化剂催化乙烯与高级 α-烯烃无规共聚得到。
从组成上看,POE 中含有较高的共聚单体( 特别是乙烯/1-辛烯的共聚物) ,导致聚合物密度较低( <0.890 g /cm3 ) ,分子中聚乙烯链段被分割为数量更多、长度更短的结晶区( 起到塑料相作用) ,用于物理交联。
另一方面,随着聚合物中长链的 α-烯烃含量的增加,长支链破坏结晶区而形成无定形的橡胶相,独特结构导致 POE 常温下拥有橡胶的髙弹性,高温下还能够塑化成型,该结构赋予 POE 优异的力学性能以及良好的抗腐蚀性和极佳的流变性能。
此外,POE 与聚烯烃材料亲 和性好,有效增强低温韧性且性价比高,因而被广泛地应用于汽车零部件、电线、电缆、机械的外包装、日常的生活用品、儿童玩具、娱乐和运动用品、密封件和热熔胶等领域。
目前我国市场对 POE 的年需求量为 2×105 t,并以 11% ~ 13%年增长率上 升. 已经实现 POE 工业化的有 Dow、Exxon、Mitsui 和 SK、LG 等多家公司,我国目前还没有 POE 生产 技术,进口 POE 产品价格居高不下 ( 15700 ~ 18800 元/t) ,使得我国 POE 应用领域受到了极大限制。
因此研发新一代聚烯烃催化剂对于我国在聚烯烃工业中的中、长期战略目标具有十分重要的意义,国内应加大对 POE 产业的支持,提高研发力度,加快 POE 国产化进程,实现 POE 的自主工业化,提升聚烯烃类产品的核心竞争力。
POE 的出现离不开过渡金属催化剂的发展,通过改变催化剂配体结构,可以准确地调控聚合物的微观结构,从而获得不同性能的 POE 产品。常用的 POE 催化剂主要包括 CGC、FI 催化剂等,我们主要对这两类催化剂近年在化学结构、催化活性等方面以及所制备聚合物的性能进行分析,并对其发展趋势予以展望。
1 限制构型催化剂( CGC)
1.1 单中心茂配体 CGC
1990 年,Shapiro 等合成第一例 CGC 并确定了其空间结构,但是没有用于烯烃聚合反应。1993 年美国 Dow 公司采用高温溶液法 Insite 工艺,通过 CGC 首次制得 POE 并实现工业化生产。
与传统的 Ziegler-Natta 催化剂相比,CGC 催化剂性能独特,可控制聚合物分子量( Mw) 、立体规整结构以及共聚单体含量,生产出窄分子量分布( MWD) 、 长链支化的高性能 POE。
将环戊二烯基( Cp) 通过一个桥基与另一个配体相连,并同时与金属中心进行配位,限制金属中心与茂环的相对旋转,因此称为 CGC,是一种桥联单茂金属结构,IV 价态的中心金属为 14 电子结构,属于缺电子体系,强 Lewis 酸可将该催化剂高效地活化为正离子,与烯烃配位后发生聚合反应。
Cp 环、过渡金属与杂原子( 例如氮) 之间( Cp-M-N) 的二面角一般小于 115°,该结构中二齿配位体限制了金属空间结构,同时桥基团的存在使配位体的位置发生偏移,从空间构型上迫使催化剂活性中心只能向一个方向打开,Suo 等专门对此进行了综述,指出开放型结构有利于大位阻 α-烯烃插入,得到具有长支链结构的聚烯烃,共聚物的加工流变性能优异,此外 CGC 催化剂一般具有良好的高温耐受性,适合于工业生产 POE。
通过对 CGC 催化剂( 如图 1) 结构进行修饰,改变桥基、Cp环上以及N原子上的取代基,或者通过催化剂组合改变催化剂催化活性以及聚合物 Mw、分子量分布( MWD) 、立体规整度等。依据桥基的不同,CGC 催化剂分为硅桥 CGC、碳桥 CGC、杂环 CGC 与其他类型 CGC。
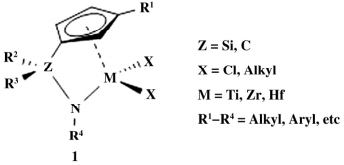
图 1 CGC 的结构
1.2 硅桥 CGC
传统 CGC 催化剂中 Si 和 N-取代基处于不同平面上,研究表明将它们合并在一个平面内能迫使催化剂的活性位点更加暴露,显著提高催化活性和 α-烯烃插入率。
2017 年,Seul 等使用 Schrock’ s Mo 催化剂通过关环复分解反应制备了一系列的氮杂硅烷钛催化剂 ( 如图 2) ,在[Ph3 C][B( C6F5 ) 4]/ i Bu3Al 助催化体系作用下进行乙烯/ 1-辛烯共聚反应( 如表 1) 。
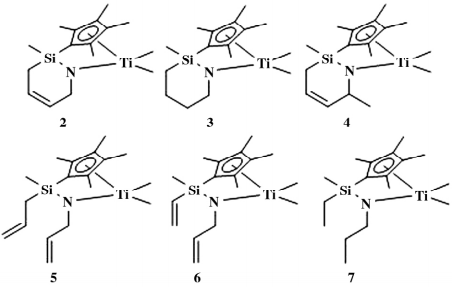
图 2 氮杂硅烷结构的 CGC
表 1 乙烯与 1-辛烯共聚结果

可以看出,该类催化剂制备的共聚物中 1-辛烯含量较高,达到 13% ~ 20% ( 摩尔分数) 。实验表明,在以甲基环己烷为溶剂、120~150 ℃ 聚合时,催化剂 3 的活性最高( 6.55 × 106 g /mol) ,聚合物 Mw 最低( 20.3×104 g /mol) ; 环状催化剂 2-4 的活性普遍高于非环催化剂 5-7,Mw 则相反,除了 3,其他环状催化剂得到聚合物 Mw 普遍低于非环催化剂。
茂金属催化剂失活机理一般认为是生成处于休眠态“[Ti( μ-R) AlR]2”双金属物质,为了抑制失活双核体形成,该小组以 ([i Bu2Al]2O) 替代( i Bu3Al) ,发现催化剂的催化活性普遍提高 1.5 到 2.7 倍。
研究结果表明,某些具有刚性配体结构的多核催化剂分子中往往存在着金属-金属的协同效应,影响催化剂的催化性能以及聚合物的微观结构。
2017 年,Liu等报道了一种新型双核钛催化剂( 如图3) 并用于烯烃的聚合反应,在rac和meso 两种构型中 meso-8 的热力学稳定性好,rac-8 在 100 ℃会发生二聚反应。使用不同剂量的[Ph3C+ B( C6F5 ) - 4]或 B( C6 F5 ) 3进行活化能得到一系列构型变化的催化剂,用于丙烯、苯乙烯和 1-辛烯的共 聚反应。
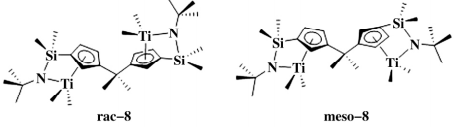
图 3 含钛的双 CGC 催化剂
meso-8 和 rac-8 在最佳的聚合条件下催化乙烯/1-辛烯共聚反应,rac-8 活性高( 3.27 × 107 g / ( mol·h·MPa) ,是同等条件下乙烯均聚反应的 1.9倍; meso-8 活性低,仅为 meso-8 的 2.77%, 它们的 1-辛烯插入率相似( ~ 7.0% ( 摩尔分数) ) , 是相应单核催化剂的两倍,作者认为可能是双金属催化剂中的第二个金属对 α-烯烃进行捕获导致。
两种催化剂均得到高支化度聚合物,支化度比相应单核 CGC 高两倍。CGC 催化剂中 N 原子上的取代基对催化剂的催化活性和共聚物的性质有重要影响。
2002 年, Nomura 课题组研究了 N 原子上取代基影响,发现环己基、叔丁基能够有效提高烯烃聚合速率。
2018 年,Williams等合成并表征了 6 种全甲基茚基钛催化剂( 如图 4) 并用于乙烯淤浆聚合反应。实验证明随着 N 上取代基给电子能力的增加( t Bu > i Pr > n Bu > 4- n Bu-Ph > 4- t Bu-Ph > Ph) 催化活性增加,其中 9( 3.60×107 g /( mol·h·MPa) ) 最高,超过经典 CGC[Me2 Si( t -Bu NCp * ) TiCl2,1. 3 × 107 g / ( mol·h·MPa) ],表明茚环催化活性更高,聚合物的熔点相差不大( 132.26 ~ 135.38 ℃ ) 。
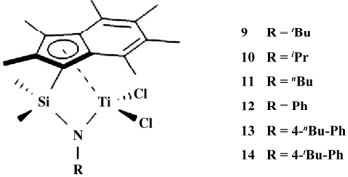
图 4 6个全甲基茚配体的 CGC 催化剂
实验还证实,增加 sMAO( solid poly-methylaluminoxane) 的量会显著降低活性,原因在于增加 sMAO 会增加载体上惰性位点,从而降低催化活性。
2018 年,Buffet 等通报了 8 种 CGC 催化剂 ( 如图 5) ,固载于 sMAO 上用于乙烯聚合反应,其作者将这 8 种催化剂分成 3 类进行讨论:
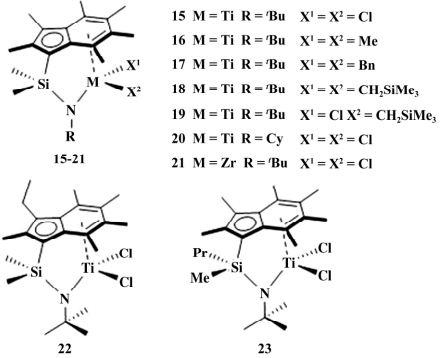
图 5 8 个具有不同取代基的 CGC
I: 离去基团 X 对乙烯聚合反应的影响: 60 ℃ 时 18 的活性最高( 7.04×107 g /( mol·h·MPa) ) ,19 的活性最低( 2.94×107 g /( mol·h·MPa) ) ; X 显著影响聚烯烃的 Mw,18 得到的聚合物的 Mw 最高 ( 1.9×106 g /mol) ,15 得到的聚合物 Mw 最低( 6.5× 105 g /mol) ,所有聚合物 MWD 均较宽( 3.5~6.0) 。
II: N 上取代基对乙烯聚合反应的影响: 15 的 活性是 20 的 5 倍,表明取代基给电子能力增加会 提高聚合活性;
III: 茚环、桥基和金属中心对乙烯聚合反应的影响: 50 ℃条件下 22 的活性( 4.07×107g /( mol·h· MPa) ) 比 15 高,证实茚上取代基对催化活性有重要影响; 与一般催化剂规律相反,金属中心为钛 ( 15) 的活性高于相应的锆( 21) 。
15、17、22、23 催化乙烯/1-己烯共聚,得到的 共聚物中 1-己烯的插入率较低( x: 1.9% ~ 2.4%) , 随着聚合温度升高( 30~90 ℃ ) ,1-己烯的插入率稍 增加( x: 1.9% ~ 2.6%) 。
同时发现 1-己烯的加入使 聚合物的 Mw 显著降低( 如 15 在 70 ℃聚合,Mw 由 4.1×105 降到 9.5×104 g /mol) . 在乙烯/苯乙烯共聚 反应中,苯乙烯的插入率较低( 1.6% ~2.5%) . 从实用效果上看,硅桥 CGC 是目前唯一应用 于商业生产的 POE 催化剂,由于受专利技术限制, 目前除了 DOW 公司,还没有其他公司使用该类催化剂生产 POE 的报道.
1.3 碳桥 CGC
2001 年,Erker 课题组报道了第一例碳桥 CGC催化剂( 如图 6) ,催化乙烯均聚和乙烯/1-辛烯共聚表现出很高的催化活性,且 1-辛烯插入率可 达 20%. 碳桥 CGC 空间构型稳定,使用 sp3 C1连接 Cp 环和 N 原子,限制了 Cp 环对中心金属的旋转, 固定了 Cp 环与中心金属原子的位置。
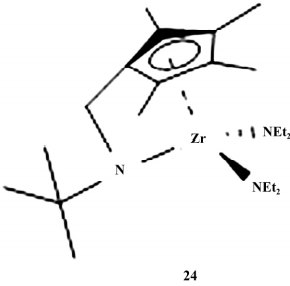
图 6 第一个碳桥 CGC
而且碳原子 半径比硅原子小,Cp-M-N 形成的咬合角较同类硅桥小 10°左右,因此碳桥 CGC 分子的空间结构更加开放,有利于大位阻 α-烯烃的插入; 另一方面,桥基是 sp3 杂化的碳,结构上位于 Cp-C-N 聚合中心的后方,完全不影响烯烃的插入,因此该类催化剂具有良好的催化活性和共聚能力。
2018 年,许课题组在前期工作的基础上合成一系列不对称 CGC 催化剂( 如图 7) ,具有不对称 的双中心结构,不仅维持了单中心催化剂较高的活 性,而且通过修饰桥基碳上取代基、调节碳桥长度,控制两个金属中心的电子环境与空间环境,调控聚合物的 MWD,得到宽分布聚烯烃。
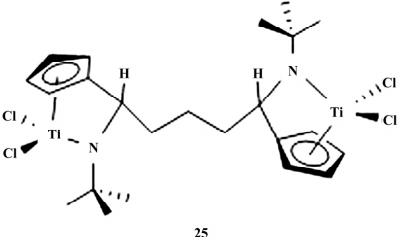
图 7 双核钛 CGC 催化剂
应用于乙烯均聚,催化活性为 1.4×106 ~ 5.7×106 g /( mol·h) ,聚乙烯的 Mw 在 1.2×104 ~ 1.9×104 g /( g·mol -1 ) , MWD 为 3.13~6.26,说明两个金属中心产生了协同作用。
用于乙烯/1-己烯共聚,发现其活性位于 1.0× 106 ~6.4×106 g /( mol·h) 之间,共聚物 Mw 显著提高( 1.6×104 ~2.9×104 g /( g·mol -1 ) ) ,MWD 变化不大,1-己烯的插入率最高可达 7.8%( 摩尔分数) 。铬系催化剂在聚烯烃领域具有重要应用,但由于铬类化合物的顺磁性,使其合成方法学研究较难进行。
2019 年,Song等制备了一系列限制构型的铬催化剂( 如图 8) 。X-ray 衍射表明,该类催化剂空间结构类似三脚钢琴凳,两个 Cl 为支脚,N 位于六 元螯合环中。
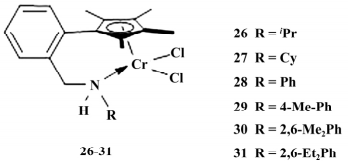
图 8 6 个铬系催化剂
在i Bu3Al /Ph3CB( C6F5 ) 4或 MAO 活化后进行乙烯的均聚和乙烯/1-己烯的共聚反应中26- 31 均表现出很高的催化活性,从高到低的顺序为 29 > 28 > 27 > 26 > 30 > 31,29 催化活性最高( 均 聚: 2.15×107 g PE /( mol·h·MPa) 。
共聚: 1.94× 107 g /( mol·h·MPa) ) ,表明 N 桥上取代基对催化活性影响很大. 比较 26-31 发现取代基位阻越大,催化活性越高,值得注意的是 30 和 31 位阻过大,活性金属中心空间过于拥挤,阻碍了烯烃的配位和插入,催化活性显著降低。
31 得到聚乙烯的 Mw 最高达到 6. 0 × 105 g /mol. 得到的聚乙烯样品熔点在 137.8 ~ 141.1 ℃ 之间,结构属于线性聚乙烯; 催化得到的乙烯/1-己烯的共聚物的熔点在 122.1 ~ 131.7 ℃,MWD 为 2.41~2.91,Mw = 6.0×104 ~ 2.0×105 g / mol) ,其中 28 得到的共聚物中 1-己烯含量最高达 到 5.97%.
2020 年,Liu 等合成以吡咯为桥的 CGC 催化剂( 如图 9) ,以MAO为助催化剂进行烯烃的聚合反应。
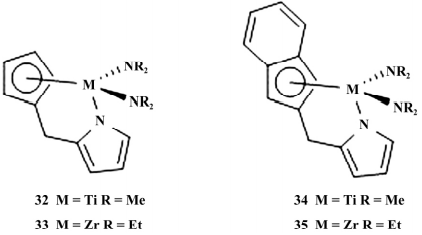
图 9 吡咯桥基 CGC
对温度、压力、铝金属摩尔比对聚合反应的影响进行了探究,使用最佳聚合条件进行乙烯/ α-烯烃共聚反应,实验表明 33 催化活性最高( 乙 烯/1-己烯: 2. 48 × 106 g /( mol·h) ; 乙烯/1-辛烯: 1.58×106 g /( mol·h) ,34 活性最低( 乙烯/1-己烯: 0.96× 106 g /( mol·h) ; 乙烯/1-辛烯: 0. 59 × 106 g / ( mol·h) ; 32 得到的共聚物 α-烯烃插入率最高( 1- 己烯为 9.81%; 1-辛烯为 8.84%) 。
这些聚合物的分子质量均较低( 1.6×104 ~3.3×104 g /mol),和一般的 CGC 催化剂不同,锆的活性高于钛,这可能是由于钛催化体系活性中心转化率较低( 52%) ,而锆催化体系的活性中心转化率较高( 80%) 。
从合成方法学上考虑,碳桥 CGC 合成方法简单; 从催化效果上看,能够达到 DOW 的经典 CGC 的水平,且国内相关研究机构掌握了自主知识产权,假以时日进行官能团优化以及工艺条件优化, 有望形成具有中国特色的 CGC 催化体系。
1.4 杂桥 CGC
将茂环与杂环( 噻吩或吡咯) 稠合得到的新型 杂环 CGC 催化剂,其活性及聚烯烃的性能有很大改善。
2017 年,Ronellenfitsch 等制备了新型喹啉基铬催化剂( 如图 10) 。在 MAO 活化下用于烯烃的聚合反应,乙烯的均聚反应中 37 的催化活性( 4.3×104 g /( mmol·MPa·h) ) 与聚合物的 Mw ( 5.5×105 g /mol) 相较于 40 提高 2 倍左右。

图 10 杂环 CGC
在乙 烯/1-己烯共聚反应中由于共聚单体效应,39 催化 活性显著提高( 2.3×103 ~ 4.1×104 g /( mmol·MPa· h) ) ,是 38 的 1.3 倍,表明三甲基硅基对催化活性 提高有效果. 36 的 1-己烯插入率最低( 4.2%) ,引入 三甲基硅基或稠合噻吩对 1-己烯的插入有促进作 用,如 38 的插入率为 5.9%,39 的插入率最高达到 9.2%。
催化剂固载在二氧化硅上,1-己烯插入率为 9.9%,Mw 为 1.0×106 g /mol。然而,杂桥 CGC 合成工艺复杂,特别是铬系催化剂因为顺磁性很难表征,原料价格昂贵,催化效果不佳,很难取代 DOW 公司 传统的 CGC 催化剂。
1.5 其他类型 CGC
2018 年,Schnee 等以苯为桥基合成了同核型茂金属催化剂和异核型茂金属催化剂( 如图 11) , 在 MAO 的活化下用于烯烃聚合反应,在乙烯的均聚反应中双核催化剂的催化活性、所得聚合物的Mw、MWD 和熔点,与单核催化剂没有区别。
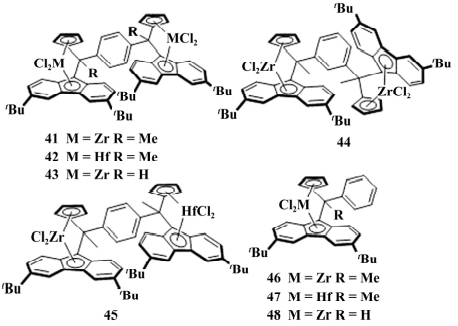
图 11 5 个双核以及 3 个单核催化剂
41 的催化活性( 24.8×106 g /( mol·h) ) 和所得聚乙烯的 Mw( 17.5×104 g /mol) 均最高,44 的 Mw 最低( 8.5× 104 g /mol) ; 含铪的 42 的催化活性最低( 6.1×106 g / ( mol·h) ) 。
43 得到的短支链聚乙烯( 甲基或少量乙基) ,可能是链行走机理。在乙烯/1-己烯的共聚反应中( 如表 2) ,双核催化剂与相应单核催化剂的 活性区别不大。
表 2 乙烯与 1-己烯共聚结果
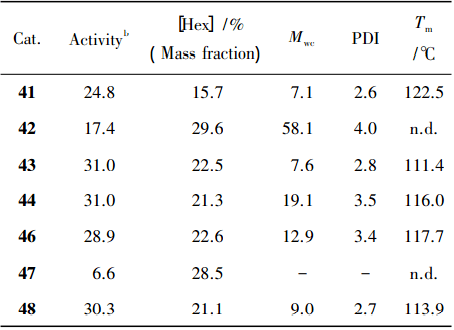
44 相对于单核催化剂得到了分子质 量更高的共聚物。Hf 类催化剂相对于 Zr 类催化剂 得到了质量更高、1-己烯插入率更高的共聚物,如 42 的催化活性( 17.4×106 g /( mol·h) ) 是 47( 6.6× 106 g /( mol·h) ) 的 2 倍,且 42 的 1-己烯含量最高 ( 29.6%) 。
2018 年,Jende 等利用富烯和 1,3,5-三( 取 代芴基) 苯合成一系列新型三核催化剂( 如图 12) , 以 MAO 为助催化剂进行烯烃聚合反应,很遗憾三核催化体系没有表现出明显的间协同作用。
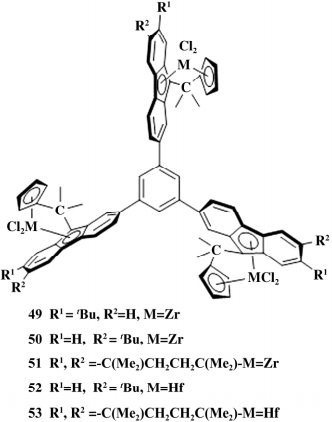
图 12 5 个三核催化剂
乙烯的均聚反应中 49 催化活性最高( 5.89 × 106 g /( mol· h) ) ,Mw 也最高( 7.1×104 ( g /mol) )。所有聚合物的熔点( 134. 3 ~ 135. 5 ℃ ) 和分子质量分布( 4. 5 ~ 5.5) 均相差不大。乙烯/1-己烯的共聚反应中 49 的 催化活性( 1.12×107 g /( mol·h) ) 是 51 的 2.7 倍,己烯插入率都不高,可能由于活性金属周围空间位 阻比较大,大位阻烯烃不易插入。
2020 年,Uborsky等为寻求工业上更有效的单中心乙烯/1-己烯共聚催化剂,制备一系列很少被研究的 C1 对称 2-取代茚基催化剂( 如图 13) 并进行 QSAR 模型检测。
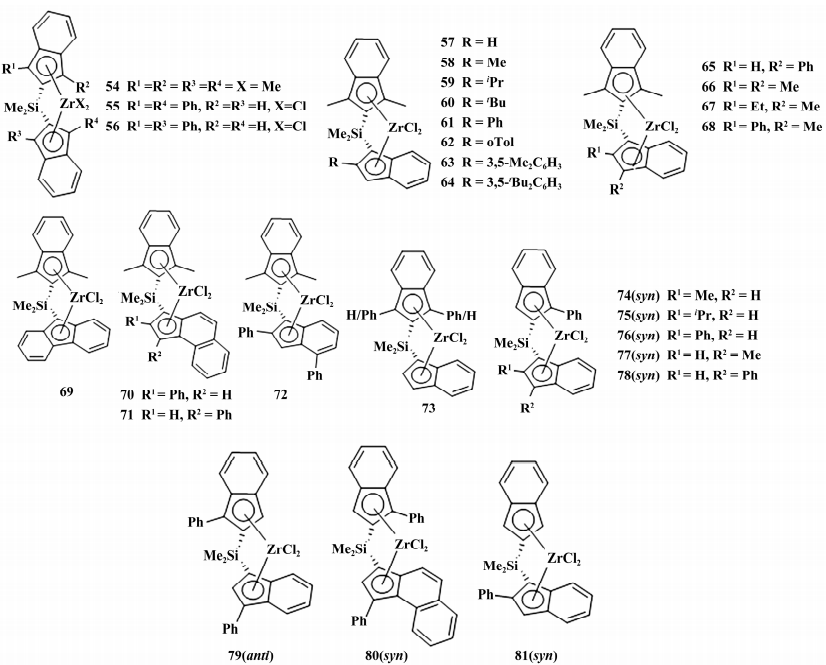
图 13 2,8-取代的茚基催化剂
在 乙 烯/1-己烯共聚反应中,54-56 的 1-己烯插入率( 0.8%~1.6%) 和聚合物分子质量( 1.5×104~8.8×104 g /mol) 均较低,其余催化剂分为两个子集讨论: 茚基片段含 1,3-二甲基为集 合 I( 57 - 72) ; 茚基片段含 2-苯基为集合 II( 73 - 80) ; 催化剂 81 介于两个子集之间。
不同点: I 型催化剂中 1-茚基片段的 2 位取代对 1-己烯插入和聚合物分子量有重要影响。对比 57,58 和 60,发现随着 R 空间位阻增加单体插入率降低( 5.1% ~2.3%) ,聚合物分子质量增加( 1.5× 104 ~7.0×104 g /mol) ; 而取代基给电子能力占主导时,1-己烯插入率和聚合物分子质量均增加,如 59 和 61 的 1-己烯插入率( 7.7% ~8.8%) 远大于 57,相对 Mw( 10.5×104 ~11.5×104 g /mol) 远大于 60。
原因在于 2-茚基片段取代基与硅甲基接近扭曲了催化剂骨架,共聚单体不易插入。可以发现 65 活性 ( 15.8×106 g /( mol·h) ) 最高,72 的 1-己烯插入率 ( 10.7%) 最高.
II 型催化剂中: 1-茚基片段上 2 位取代基对共聚单体的亲和力影响较小,随着取代基位阻增加 74-76 的 1-己烯插入率变化不大( 4. 2% ~ 5.2%) ,而聚合物 Mw 显著下降( 15.9×104 ~ 10.5× 104 g /mol) 。对比催化剂 76 和 81,可以发现引入苯基对 1-己烯插入率和聚合物分子质量都有负面作用。
对比 61 和 65( 或 70 和 71,74 和 77) ,发现将 2位取代基移到3位上,1-己烯插入率和聚合物分子质量均降低; 将茚替换为萘并茂环,如 61 和 70 ( 或 65 和 71,78 和 80) ,1-己烯插入率稍微降低,聚合物 Mw 显著升高。我们认为上述催化剂体系合成路线长,结构复杂,催化效果尚且不能达到 DOW 公司经典 CGC 水平,工业上很难大规模推广应用.
2 FI 催化剂
2.1 FI 催化剂
1995 年,Cozzi 课题组报道,使用羟苯基噁唑啉配体与过渡金属键合形成配合物,可以作为乙烯聚合催化剂,但催化活性较低。1987 到 2001 年间 Fujita 小组开发了苯氧基亚胺配体的 IVB 族烯烃聚合催化剂,现在被称为 FI 催化剂( 如图 14) 。
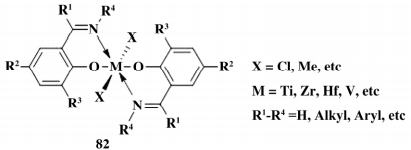
图 14 FI 催化剂结构
FI 催化剂作为非茂金属催化剂,依靠配体分子内 O,N 等杂原子与金属中心键合,又被称为[NO]型催化剂,Liu 等对 II 型[NO]催化剂进行了综 述,此外 IF 型、PI 型和 FE 型催化剂也都迅速发展起来。
FI 催化剂使用的助催化剂,有 MAO、异丁基鋁/苯基硼以及氯化镁负载的异丁基鋁等,该类催化体系活性高,可以达到 10 倍茂金属,应用于制备低分子量聚乙烯、超高分子量聚乙烯、乙烯/α-烯烃共聚物等。
FI 催化剂配体修饰比较灵活,影响后续的烯烃聚合。该类催化剂可实现多重活性聚合,对此进行的综述,揭示 了各类取代基对性能的影响。
2.2 FI 催化剂的进展
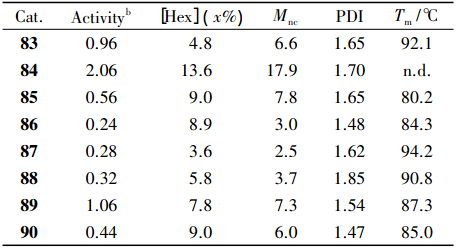
2017 年,Li 课题组通过修饰 β-酮亚胺配体合成水杨醛亚胺和 β-酮亚胺杂配钛催化剂( 如图 15) ,在MMAO的活化下用于催化烯烃聚合。在乙烯的均聚反应中,86 的催化活性是同类型 β-二酮亚胺催化剂的 10 倍。比较催化活性发现 83 > 84 > 85,表明取代基 R2 的性质对催化活性有重要影响。
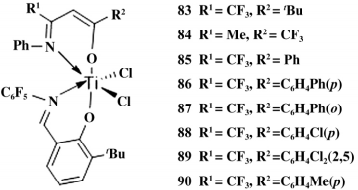
图 15 β-二亚胺类型 FI 催化剂
在乙烯/1-己烯的共聚反应中( 如表 3) ,84 由于取代基位阻最小,活性金属中心空间大,因而催化活性高( 2.06×107 g /( mol·h·MPa) 、1-己烯插入率高 ( 1-己烯浓度为 2.0 mol /L 时,插入率为13.6%) ,聚 合物 Mw 较高( 17.9×104 g /mol) 。利用双金属催化剂的协同效应可以有效解决单金属催化剂得到的聚合物分子量分布较窄这个问题。
表 3 乙烯与 1-己烯共聚结果
2018 年,Wang 等合成了对苯基桥联双金属 FI 催化剂( 如图 16) ,这类双金属催化剂保持了单中心催化剂的活性,可以很方便的通过调节取代基控制两个金属中心的电子环境与空间环境,进而调控聚合物的 MWD。
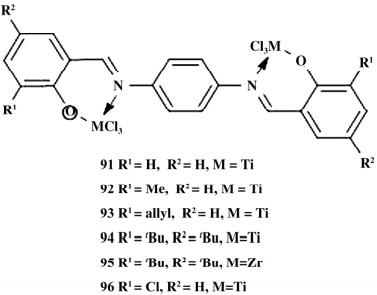
图 16 苯基桥连双核催化剂
以 MAO 为助催化剂催化乙烯的聚合反应,制备出催化活性较高( 2. 9 × 105 g / ( mol·h) ) 、MWD 较宽的聚乙烯。在乙烯/1-己烯的共聚反应中,在亚胺镍和乙基锌组成的催化体系里,1-辛烯插入率( 1.06% ~ 13.63%) 较高,聚合物结晶度低于 50%。
2006 年 Arriola 等首次提出链穿梭聚合法,利用锆基 FI 催化剂与铪基催化剂在链穿梭剂二乙 基锌作用下制备具有交替半结晶和无定型链段结构 的烯烃嵌段共聚物( OBC) 。
2017 年,Zhang 等合成了一种新型非氟代的双核FI催化剂( 如图17) ,以 MAO 为助催化剂,用于烯烃的聚合反应,乙烯的均聚反应中催化剂活性较高( 2.0×106 g /( mol· h) ) ,MWD 较窄 ( Mn : 9. 8 × 104 g /mol; Mw /Mn : 1.79) ,与 α-二亚胺镍( Ⅱ) 催化剂构成多组分催化体系,在链穿梭剂二乙基锌作用下催化乙烯与 1-辛 烯共聚,发现 1-辛烯插入率为 4.9%,分析发现聚合 物“软段”部分主要由乙烯/1-辛烯共聚物构成,“硬 段”部分主要由线性聚乙烯构成,DSC 结果显示为双峰,Tm分别为 91.32 和 126.61 ℃,表明其为 OBC。
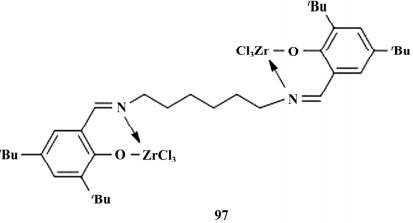
图 17 双核 FI 催化剂
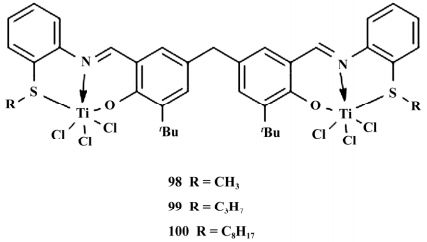
图 18 三齿配体[ONS]双核钛催化剂
2017 年,Xie 课题组报道了首例非茂三齿双核钛催化剂,在 MMAO 的活化下,能够高活性地催化乙烯聚合以及乙烯与 α-烯烃共聚,得到 Mw 相对较高、MWD 相对较窄的聚合物。
研究发现双核的催化活性以及聚合物的共聚单体插入率远高于相应单核的。在 25 ~55 ℃条件下随着温度的升高,100 的催化活性增大,聚合物的 Mw 增加,而随着 Al ∶ Ti 比值的增加,100 的催化活性增加。
最佳反应条件为 55 ℃,Al ∶ Ti = 1500,在相同的条件下催化活性降低次序为 100 > 99 > 98,这表明取代基中烷基链越长,在溶剂中溶解性能越好,催化活性就会越高。
反应中,98 甲硫基位阻最小,催化乙烯/α-烯烃的 共聚 活 性 ( 乙 烯/1-己 烯: 2. 52 × 107 g /( mol · h · MPa) ; 乙 烯/1-辛 烯: 2. 94 × 107 g /( mol · h · MPa) ) ,其 单 体 插 入 率 ( 1-己 烯: 9. 3%; 1-辛 烯: 9.8%) 也是最高的。
为了提高其催化性能,研究者们尝试将茂环和苯氧基亚胺同时引入到一个分 子中。2002 年,Huang 课题组首次合成该类催化剂,催化剂活性大为提高。
2018 年,Invergo 等合成一种 FI-茂混合配体的单/双金属催化剂( 如图 19) ,研究发现在乙烯的聚合反应中 102 得到的聚乙烯分子量( 1.8× 105 g /mol) 是同类型 Cp* TiMe3的两倍,而 101 只能得到分子质量较低的聚乙烯( 4.4×104 g /mol) ,这表明双金属催化剂具有协同效应,通过与聚合物 C— H 键之间的元节效应,稳定了聚合物的链增长,同时抑制链终止。
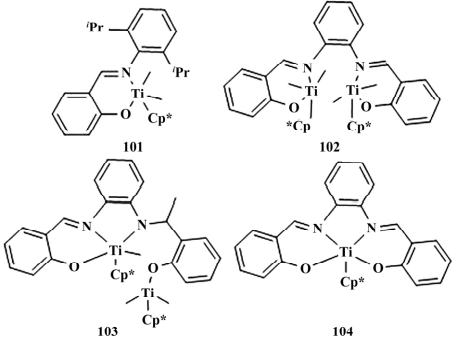
图 19 茂环与苯氧亚胺混合型 FI 催化剂
104 对乙烯/1-己烯的共聚反应没有催化活性。作者认为当存在 1-己烯时 102 快速裂解 成 104 和 Cp* TiMe+ 2,因此 102 产生的聚合物的 Mw ( 9.8 × 104 g /mol) 和 1-己烯的插入率( 18. 2%) 均与 Cp* TiMe3十分接近。
该研究成果表明双金属催化剂中金属-金属间距离过于接近会限制金属间的协 同效应,导致催化活性降低。近几年来,M-Cl 和 M-alkane 型催化剂得到了 发展,相比较而言 M-NR2 型催化剂没有受到重视。
2019 年,Gao 等创造性地制备了一系列 FI 型锆 胺 催 化 剂 ( 如 图 20 ) ,并依次用三甲基铝 ( TMA) 和[Ph3C][B( C6F5 ) 4]活化后进行乙烯均聚 和乙烯/1-辛烯共聚反应。

图 20 烷基氨类型 FI 催化剂
在相同条件下,二甲胺型105 的催化活性较低为 2. 85 × 107 g /( mol · h · MPa) ,而 1-辛烯插入率最高( 7.2%) ; 苄基型 106 的催化活性最高达到 5.65×106 g /( mol·h) ,而 1- 辛烯插入率仅为 1.0%; 甲基型 107 的催化活性中等,1-辛烯插入率最低( 0.9%) ; 相应的单 FI 型 109 的催化活性最低为 1.81×106 g /( mol·h) ,1-辛烯插入率较高 ( 6. 3%) ; 108 没有活性; 所有聚合物 MWD 都比较窄( < 2.9) 。
研究机理表明,聚合链从 锆( 或钛) 转移到铝是催化剂失活的主要途径,而原 位生成的单 FI-Zr 结构可能是导致这种独特催化性 能的主要原因,该单 FI-Zr 结构通常无法通过烷基或氯化物前体获得,只能由 M-NR2型催化剂获得。
FI 催化剂因为合成方法简单,容易修饰且催化活性高,这些年得到了迅猛发展,更为关键的是 FI 催化剂可以使用烷基铝作为助催化剂,基于价格考虑,大大降低了催化成本,非常有希望在烯烃共聚 领域取得工业化成果。
3 结论与展望
聚烯烃行业竞争的核心是催化剂的设计、合成与应用,POE 产业是聚烯烃材料中最有市场价值的部分,关乎我国石油化工下游产业升级换代,国内外的相关研究者对此进行了长期的研究,我们总结了近年来 POE 催化剂的研究进展,针对如何提高 α-己烯( 或 1-辛烯) 插入率、控制产品分子量和分子质量分布进行了讨论。
传统采用分步法合成茂金属催化剂,这类方法合成路线长、步骤多、合成产率低( <50%) ,近几年研究表明,通过改变起始原料及路线,可以简化合成步骤,避免中间产物的分离纯化,提升催化剂的合成产率,同时经过配体改良,可获得工业所需的耐高温催化剂。


目前评论: